El sistema inmune se compone de varios tipos de células muy especializadas, encargadas de los procesos de inmunidad celular y humoral. Estas células son los linfocitos (linfocitos T y B, y linfocitos no T no B), las células plasmáticas y los macrófagos, casi todas ellas tienen su origen en la médula osea; sin embargo, durante la vida fetal y neonatal los linfocitos sufren un proceso de condicionamiento en órganos linfoides centrales, como el timo.
Una vez terminado el proceso de diferenciación, estas células pasan a los órganos linfoides periféricos, por ejemplo los ganglios linfaticos y el bazo.
LINFADENOPATIA
Estructura y función de los ganglios linfáticos
Los ganglios linfáticos se distribuyen en grupos, a lo largo de los vasos linfáticos de todo el organismo. Tienen forma ovoide y su tamaño varía normalmente desde unos pocos milímetros hasta más de 1 cm. Su estructura facilita la filtración eficaz de la linfa y la migración interna de células, sobre todo de los linfocitos y los macrófagos.
Los ganglios linfáticos, cuentan con una malla de fibras reticulares que sirven de soporte y organización para sus elementos. Por otra parte, la red fibrilar sirve también para crear un sistema de canales a través de los cuales fluye la linfa, y forma una densa cápsula en la superficie del ganglio.
Los vasos linfáticos aferentes entran en el ganglio perforando la superficie de la cápsula, y drenan en el seno subcapsular. Desde allí, la linfa fluye hacia el interior del ganglio a lo largo de los canales formados por la estructura reticular, y sale de él por el hilio a través de un único vaso linfático eferente. La sangre entra al ganglio a través del hilio por una pequeña arteriola.
Los capilares arteriolares están en conexión con las vénulas poscapilares, que a su vez están tapizadas por un endotelio característico, formado por células cuboides altas. Los linfocitos pasan entre estas células endoteliales para alcanzar la malla reticular del ganglio.
El ganglio linfático consta de tres zonas anatómicas. En la corteza, junto al seno subcapsular, hay agregados de linfocitos B, denominados folículos linfoides. Algunos folículos contienen centros germinales, con células plasmáticas, macrófagos y linfocitos de rápida división, ocupados activamente en la síntesis de proteinas. Entre los folículos y junto a ellos está la zona paracortical, que consta de varias capas de linfocitos T. Debajo del paracortex y ocupando la parte central del ganglio, se encuentra la médula, donde los linfocitos se ordenan en forma de cadenas, denominadas cordones medulares, que convergen en el hilio.
RESPUESTA DE LOS GANGLIOS LINFATICOS A LA AGRESION ANTIGENICA
Los antígenos llegan a los ganglios linfáticos a través de los vasos linfáticos aferentes, y son englobados y procesados por los macrófagos corticales. Dentro del ganglio el antígeno se concentra en la zona de contacto entre el paracórtex y los folículos linfoides, lugar en el cual los linfocitos T y B están en estrecha yuxtaposición. El ganglio linfático sirve como punto de reunión de todos los elementos necesarios para que se inicie la respuesta inmune.

Ganglio linfático. En la superficie del ganglio las fibras reticulares forman una cápsula fibrosa densa, los vasos linfáticos aferentes penetran perforando la superficie y drenan en los senos subcapsulares. De allí la linfa fluye al interior por canales formados por la malla reticular y sale del ganglio por el hilio a través de un vaso linfático eferente único. Los folículos linfáticos están constituidos por los linfocitos B, en los centros germinales hay macrófagos, células plasmáticas y linfocitos en rápida división. Entre los folículos y adyacentes a ellos, está la zona paracortical de los linfocitos T
Pacientes con linfadenopatía: Consideraciones generales.
Edad: En pacientes con adenopatías se espera que un 20 % de las lesiones sean malignas en menores de 30 años, mientras que en los mayores de 50 años la proporción de adenopatías malignas es del 60%.(Jay H. Stein)
Determinar si la adenopatía es localizada o generalizada y conocer el proceso cronológico de su desarrollo.
En ciertas regiones del organismo, el trastorno tiene una significación clínica especial. Por ejemplo, el hecho de que los ganglios supraclaviculares sean palpables indica la posibilidad de que exista un proceso maligno intratorácico o intraabdominal y por lo tanto, requiere una evaluación cuidadosa. Por el contrario, la linfadenopatía occipital aislada raras veces es manifestación de un proceso maligno, sino más bien de una infección del cuero cabelludo como la tiña o las producidas por picaduras de insectos.
El aumento progresivo del tamaño de los ganglios linfáticos durante varias semanas, especialmente si se asocia a fiebre, escalofríos, sudoración nocturna o pérdida de peso, puede indicar una enfermedad sistémica grave, tal como una infección crónica por micobacterias u hongos, o un proceso linfoproliferativo maligno.
Los ganglios linfáticos dolorosos son sugestivos de los procesos infecciosos, pero no separan adecuadamente los procesos malignos de los benignos. Por ejemplo, el rápido aumento de tamaño de los ganglios linfáticos en la leucemia linfoblástica aguda puede asociarse a malestar considerable. Un síntoma peculiar, descrito en algunos casos de la enfermedad de Hodgkin, es el dolor referido a los ganglios linfáticos agrandados, tras la ingestion de bebidas alcoholicas.
La consistencia
Cuando son dolorosos a la presión, calientes y eritematosos, con fluctuación o estrías linfangíticas en la piel adyacente reflejan procesos infecciosos locales. Los ganglios duros, de consistencia petrea, fijos a los tejidos adyacentes, indican la posibilidad de un proceso maligno, especialmente de un carcinoma o sarcoma metastásico. En cambio, los ganglios móviles, de consistencia elástica, son propios de los linfomas.
Linfadenopatía regional
Ganglios linfáticos cervicales: Reciben el drenaje linfático procedente de la cabeza, cuello y cavidad orofaringea, es posible considerar como causa del trastorno las infecciones de tejidos blandos de cara, los abscesos dentarios, las otitis externas y la faringitis bacteriana. La mononucleosis infecciosa puede presentarse como linfadenopatía cervical localizada y su diagnóstico requiere la realización de pruebas serológicas específicas y un examen de los extendidos de sangre periférica.
Entre los procesos malignos que también pueden expresarse por una linfadenopatía localizada cervical, se incluyen la enfermedad de Hodgkin, los linfomas no Hodgkin y los carcinomas espinocelulares de estructuras nasofaríngeas y laríngeas.
Ganglios linfáticos axilares. Los ganglios de las axilas drenan los vasos linfáticos de las extremidades superiores y de las mamas. La adenopatía axilar debe sugerir procesos infecciosos como la fiebre por arañazo de gato, la esporotricosis, la tularemia y las infecciones estafilocócicas o estreptocócicas
El examen de las extremidades superiores en busca de picaduras, lesiones supurativas o linfangitis puede proporcionar datos importantes para el diagnóstico.Entre los procesos malignos que se acompañan de adenopatía axilar localizada cabe citar los linfomas, los melanomas y los carcinomas de mama.
Ganglios linfáticos epitrocleares. La linfadenopatía epitroclear bilateral indolora que se presenta en trabajadores manuales, puede ser el resultado de pequeños traumatismos repetidos, de infecciones, o de ambos. Su aparición en otras circunstancias es un indicio de linfoma.
Ganglios linfáticos supraclaviculares. La palpación de adenopatías supraclaviculares es un signo de mal pronostico, pues dada su frecuente asociación, habra que considerar
siempre la posible presencia de un proceso maligno intratorácico o intraabdominal. El drenaje linfático del torax y del mediastino se dirige bilateralmente hacia dichos ganglios. El conducto torácico que lleva el drenaje linfático abdominal, drena en el confluente yúgulo-subclavio, en la región supraclavicular izquierda. Este hecho anatómico explica el fenómeno bien conocido según el cual el ganglio supraclavicular izquierdo (ganglio de Virchow) actua como centinela para anunciar la presencia de una neoplasia abdominal no detectada. Las infecciones intratorácicas crónicas, generalmente por micobacterias o micóticas, pueden presentarse también asociadas a linfadenopatía supraclavicular localizada, lo mismo que la sarcoidosis. Las infecciones bronquiales o las neumonías bacterianas, sin embargo, no se manifiestan de este modo. Para establecer el diagnóstico definitivo en casos de aumento de tamaño de los ganglios supraclaviculares, está indicada la biopsia precoz de una de las adenopatías.
Ganglios linfáticos inguinales. Es muy dificil de evaluar, ya que virtualmente en todos los adultos se observa cierto aumento del tamaño de estos ganglios, como consecuencia de infecciones menores del aparato genital y las extremidades inferiores.
Por los ganglios inguinales pasa el drenaje linfático de las extremidades inferiores, de la piel de la mitad inferior del abdomen, así como de los genitales y del periné.
Es importante recordar que los organos pelvianos internos y los testículos drenan a través de los ganglios ilíacos a la cadena para-aórtica, de modo que las infecciones o procesos malignos pélvicos profundos no producen casi nunca linfadenopatía inguinal. Las posibilidades diagnósticas a considerar en estos casos son la celulitis de las extremidades inferiores y las infecciones venereas, como la sífilis, el chancro blando, el herpes genital o el linfogranuloma venéreo. Entre los posibles procesos malignos, cabe citar: los linfomas, las metástasis de melanomas de las extremidades inferiores y los carcinomas espinocelulares de localización primaria en pene o vulva.
Ganglios linfáticos internos. La linfadenopatía interna puede llamar la atención del médico en los exámenes radiológicos; por ejemplo, se puede tratar de una adenopatía hiliar o mediastínica en una radiografia de tórax. Indica la posible presencia de un carcinoma broncogénico o de un linfoma. La enfermedad de Hodgkin se asocia con más frecuencia a linfadenopatía hiliar o mediastínica que los otros tipos de linfoma. La sarcoidosis, la tuberculosis y las infecciones micóticas son algunas de las
enfermedades no cancerosas que deben tenerse en cuenta en el diagnóstico diferencial.
La linfadenopatía intraabdominal se detecta en ocasiones al encontrar una masa palpable en la exploración fisica o, indirectamente, por los efectos obstructivos o de compresión sobre órganos adyacentes como, por ejemplo, los uréteres.
Es necesario a veces emplear diversos tipos de técnicas radiológicas para detectar y determinar la extensión de la linfopatía intra-abdominal entre ellas figuran la pielografía intravenosa, la linfografía, la ecografía y la tomografía axial computada, actualmente se agrega la Resonancia Magnética Nuclear.
Si la linfadenopatía está limitada al abdomen y no se dispone de tejidos periféricos para su estudio, puede estar indicada la laparotomía exploradora. La linfadenopatía intraabdominal indica a menudo la presencia de un proceso maligno, que puede ser la enfermedad de Hodgkin u otro tipo de linfoma. La enfermedad de Hodgkin afecta por lo general los ganglios pélvicos y retroperitoneales, y suele respetar los ganglios mesentéricos. Por el contrario, los linfomas no Hodgkin con frecuencia afectan también dichos ganglios. En la linfadenitis mesentérica tuberculosa, a veces se observan ganglios linfáticos abdominales de gran tamaño. La linfadenopatía umbilical aislada indica generalmente la presencia de un adenocarcinoma gástrico.
Ganglios superficiales. Los pacientes con distintos trastornos dermatológicos, especialmente la dermatitis exfoliativa, tienen a veces linfadenopatías superficiales regionales, que se resuelven al mejorar la enfermedad dérmica. En la biopsia, los ganglios afectados se caracterizan por la presencia de un gran número de células reticulares atípicas y de células espumosas con sustancias lipoides o melanina.
Linfadenopatía generalizada
En el adulto, la linfadenopatía generalizada indica por lo general la presencia de una enfermedad sistémica grave, cuya naturaleza puede ser infecciosa, inmunológica o maligna. Suele estar indicada la biopsia de uno de los ganglios afectados.
Debe descartarse como posible causa la ingestión de algunos fármacos, como las hidantoinas, la hidralacina y el allopurinol, los pacientes pueden presentar fiebre, exantema, linfadenopatía, hepatoesplenomegalia, artritis e ictericia, signos que desaparecen al interrumpir el fármaco, los hallazgos anatomopatológicos en estos ganglios hacen aplicable el término seudolinfoma.
Las adenopatías generalizadas pueden ser consecuencia de una afección sistémica. Entre ellas las más frecuentes son los síndromes de mononucleosis infecciosa, como el causado por el virus Epstein-Barr, el citomegalovirus o el toxoplasma. Por otra parte, la linfadenopatía generalizada no es frecuente en los adultos con infecciones, excepto en los que padecen tuberculosis, infecciones micóticas (como histoplasmosis o coccidioidomicosis), brucelosis, endocarditis bacterianas, hepatitis infecciosa o sífilis secundaria.
Los trastornos inmunológicos que se deben tener en cuenta al evaluar un caso de adenopatía generalizada son: la sarcoidosis, la artritis reumatoide y el lupus eritematoso sistémico
Recientemente se ha descrito un trastorno denominado linfadenopatía angioinmunoblástica (LAID), que se encuentra en el límite entre las reacciones de hipersénsibilidad inmunológica y los procesos linfoproliferativos malignos.
Algunos pacientes desarrollan un verdadero linfoma maligno, que suele además ser rebelde al tratamiento.
Los procesos malignos asociados a la linfadenopatía generalizada incluyen leucemias (especialmente la linfoblástica aguda en los niños y la linfática crónica en los adultos de edad avanzada) y los linfomas.
Biopsia de los ganglios linfáticos
La biopsia de los ganglios linfáticos es en muchos casos la técnica definitiva que establece o confirma un diagnóstico, debe realizarse sin retraso en todo paciente que padezca una linfadenopatía no atribuible a una causa evidente, como la mononucleosis infecciosa, o algún foco infeccioso localizado, y cuya duración sea de una semana o más.
Es importante elegir el más representativo, en una zona donde no exista otro proceso que enmascare el cuadro histológico. Por ejemplo los ganglios de la región inguinal, femoral o cervical superior no suelen resultar útiles ya que a menudo presentan una hiperplasia reactiva, causada por procesos infecciosos localizados repetidos.
Es también importante que la cápsula del ganglio esté intacta, especialmente si se estudia la posibilidad de un linfoma, ya que las características estructurales necesarias para hacer el diagnóstico específico y la clasificación del linfoma, así lo requieren.
La afectación extraganglionar de los linfomas puede ser también diagnosticada por biopsia.
Las biopsias de los ganglios linfáticos por punción, rara vez son útiles y deben evitarse.
Al hacer la biopsia, es preciso conservar una parte de la muestra en formol para el examen histopatológico habitual.
Se debe obtener también material adecuado para llevar a cabo los correspondientes cultivos bacterianos, micológicos y para micobacterias, así como para las tinciones especiales de estos tipos de microorganismos y, en ciertas circunstancias, para tinciones citoquímicas especiales y para el estudio de los marcadores linfocíticos de superficie.
En algunos casos, como en los procesos malignos indiferenciados, puede servir de ayuda la microscopía electrónica y es necesario conservar material en soluciones fijadoras especiales para este fin.
ENFERMEDAD DE HODGKIN Y LINFOMAS NO HODGKIN
Enfermedad de Hodgkin
Etiología
La causa de la enfermedad se desconoce, se postula como posible, la trasmisión horizontal de un agente del tipo de un virus de baja capacidad infectiva y con largo período de latencia.
Se encuentra un aumento de los títulos de anticuerpos contra el virus de Epstein y Barr, aunque el aumento de la infección por este virus podría ser consecuencia de los trastornos inmunológicos de estos pacientes y no su causa.
La posibilidad de una predisposición genética o de una exposición a un agente ambiental, no se descarta, ya que existen brotes familiares de la enfermedad, y el riesgo de padecerla se incrementa de 3 a 7 veces en hermanos gemelos o familiares cercanos de los pacientes que la padecen, con respecto a la población general.
Dato adicional a favor de factores hereditarios, es que la enfermedad de Hodgkin se asocia con ciertos antígenos HLA (Complejo mayor de histocompatibilidad)
Histología
La enfermedad de Hodgkin suele aparecer en los ganglios linfáticos, que presentan entonces características de malignidad, con pérdida de la arquitectura y de la integridad de la cápsula. La celularidad normal queda reemplazada por una población heterogenea. donde existen células linfoides «histiocitarias», acompañadas de una infiltración de linfocitos, células plasmáticas y a veces eosinófilos; componentes invariables de este cuadro son las células de Reed-Sternberg, las células de Hodgkin o ambas.
Se considera que la enfermedad de Hodgkin es un proceso maligno a causa de la pérdida de la arquitectura ganglionar, de su capacidad para invadir tejidos y para metastatizar, de su letalidad potencial y de la forma en que responde al tratamiento. La célula maligna es presumiblemente la célula de Reed-Sternberg, en base a su aneuploidía, sus características de crecimiento en cultivo de tejidos y su heterotransplantabilidad al ratón atímico. Es una célula grande bi o multinucleada. con nucléolo prominente (frecuentemente con ojos de buho o imagen en espejo) un halo claro perinucleolar y una condensación de la cromatina en la perifería del núcleo. Tiene además, capacidad para dividirse. La célula de Hodgkin es el equivalente mononucleado de la célula de Reed Sternberg, con sus mismas características nucleares y nucleolares.
Histológicamente la enfermedad de Hodgkin se divide en cuatro categorías principales:
- Predominio Linfocitario
- Esclerosis Nodular
- Celularidad Mixta
- Depleción Linfocitaria
Cuando se toman biopsias repetidas en el curso de la enfermedad, se puede constatar la progresión de la enfermedad desde el predominio linfocitario o celularidad mixta, hasta la depleción linfocitaria.
Características clínicas
Le enfermedad de Hodgkin suele aparecer en adultos jóvenes (15-34 años), pero puede hacerlo a cualquier edad, con un pequeño segundo pico a partir de los 50 años. En los pacientes jóvenes predomina la forma de esclerosis nodular, mientras que en los de más edad aumenta la proporción del tipo de celularidad mixta.
En conjunto existe predilección por los varones, pero entre los 15 y 34 años la proporción de sexos es similar, y la forma de esclerosis nodular afecta en mayor medida a las mujeres.
Las adenopatías son el signo más frecuente en la enfermedad de Hodgkin. Suelen ser periféricas, y comienzan sobre todo en el cuello y la región supraclavicular, siendo mucho más rara su aparición en otros lugares como la región epitroclear o poplítea.
Las adenopatías son de consistencia firme, elásticas y redondeadas; en ocasiones pueden ser dolorosas y presentar fluctuaciones de tamaño espontáneas.
Cuando se observan adenopatías fijas y pétreas, la causa suele ser una metástasis de un carcinoma más que un linfoma o una enfermedad de Hodgkin.
En los pacientes con adenopatías debe practicarse siempre una biopsia ganglionar cuando se den las siguientes circunstancias: que las adenopatías sean grandes (>2 cm), que estén limitadas a una región anatómica, o que se localicen en lugares anormales, ej., supraclaviculares o femorales y persistan sin explicación por un proceso inflamatorio local o sistémico.
Clasificación morfológica de la enfermedad de Hodgkin
| Predominio linfocitario Linfocitos maduros con algunos «histiocitos» (Linfocitos transformados) y escasas células de Hodgkin y de Reed Sternberg Constituye el 10 al 15% de los casos Suele ser un estadío temprano de la enfermedad No suele aparecer por encima de los 50 años |
| Esclerosis nodular Nódulos de tejido linfoide separados por anchas bandas de tejido colágeno fibroso Pueden estar presentes células lacunares tras la fijación en formol Pueden no verse células de Reed-Sternberg Constituye el 20-50 % de los casos Incidencia máxima entre los 10-30 años, pero puede aparecer en cualquier edad Predominio en mujeres Suele ser un estadío temprano de la enfermedad Suele aparecer como adenopatías supraclaviculares, mediastínicas o ambas |
| Celularidad mixta Infiltrado celular pleomorfo (linfocitos, células plasmáticas, eosinófilos) con abundantes células de Reed-Sternberg y de Hodgkin Constituye el 20-40 % de los casos Incidencia máxima entre los 20-40 años Suele ser un estadío avanzado de la enfermedad Predominio en varones |
| Depleción linfocitaria Pocos linfocitos maduros; predominan los histiocitos con fibrosis y necrosis Frecuentes células de Reed-Sternberg y de Hodgkin Constituye el 5-15 % de los casos Suele aparecer por encima de los 25 años Generalmente es un estadío avanzado |
Las adenopatías centrales también son frecuentes en la enfermedad de Hodgkin, sobre todo las mediastínicas o de los ganglios hiliares del pulmón; pueden asociarse a tos, dolor torácico, o más rara vez a un sindrome de obstrucción de la vena cava superior.
La afectación abdominal puede cursar con dolor, masa tumoral o edema de extremidades inferiores; puede haber esplenomegalia e hiperesplenismo.
La infiltración de la cadena mamaria interna es rara, aunque suele ser asintomática, puede dar lugar a dolor torácico.
La forma de comienzo con afectación visceral es poco frecuente. Menos del 1 % de los pacientes debutan con lesión exclusivamente extraganglionar; aproximadamente el 10 % muestran desde el principio una invasión visceral generalmente el pulmón.
10 % tienen al descubrir la enfermedad una amplia diseminación sistémica, siendo los órganos más comúnmente afectados, el pulmón la pleura, el hueso y el hígado. En general la enfermedad comienza en órganos o ganglios supradiafragmáticos, pero en los pacientes de edad avanzada, existe cierta predilección por la localización infradiafragmática.
La enfermedad suele presentarse con síntomas generales, como fiebre en agujas, sudoración nocturna o pérdida de peso; éstos son los llamados síntomas B. Pueden aparecer: debilidad, cansancio y prurito. En ocasiones se observa un síndrome nefrótico acompañante. A veces existe dolor en los lugares afectados tras la ingesta de alcohol.
Hay en estos pacientes una gran facilidad para las infecciones por oportunistas, como la criptococosis, la infección por citomegalovirus o por Pneumocystis carinii, la toxoplasmosis, la nocardiosis, la aspergilosis, la listeriosis, la candidiasis y la tuberculosis; el herpes zoster es también frecuente. Los pacientes esplenectomizados presentan facilidad para las septicemias abrumadoras, especialmente por neumococo o meningococo.
La historia natural de un enfermo no tratado refleja una progresiva diseminación de la enfermedad, primero hacia los ganglios contiguos y después hacia las vísceras acompañada de fiebre y caquexia produciéndose la muerte, por fallo orgánico o por una infección intercurrente.
Con los tratamientos actuales la supervivencia media es mayor de 10 años, cualquiera que sea el estadio, la supervivencia a los 5 años es aproximadamente del 80 %, y a los 10 años del 60 %.
Analítica
Cuando la enfermeded de Hodgkin está en actividad, existe velocidad de sedimentación elevada, aumento de la tasa de fosfatasa alcalina leucocitaria, hiperuricemia y neutrofilia, aunque estos datos no son específicos ni guardan una relación estrecha con el avance de la enfermedad.
La afectación hepática se traduce en un aumento de la fosfatasa alcalina, de la bilirrubina, de la SGOT y SGPT, y la afectación ósea produce un incremento selectivo de la fosfatasa alcalina. En raras ocasiones puede aparecer un síndrome nefrótico con hipoalbuminemia.
Otras veces se encuentra anemia. a veces con prueba de Coombs positiva.
Una fórmula leucoeritroblástica en sangre periférica puede reflejar afectación de
la médula osea. La eosinofilia y linfopenia son frecuentes
Cuando la enfermedad está avanzada suelen aparecer deficiencias inmunológicas.
Diagnóstico diferencial
La enfermedad de Hodgkin debe diferenciarse de: Linfomas no Hodgkin, que tienen como datos diferenciales:
- Habitual mayor edad
- Tendencia a evitar mediastino y tórax
- Mayor frecuencia de afectación primaria extraganglionar.
Debe diferenciarse también de:
- Mononucleosis infecciosa
- Infección por citomegalovirus
- Tuberculosis
- Histoplasmosis
- Coccidioidomicosis
- Adenopatías por hidantoina
- Toxoplasmosis
- Sarcoidosis
DETERMINACION DEL ESTADIO DE LA ENFERMEDAD DE HODGKIN
| Estadio | Descripción |
| I | Afectación de los ganglios linfáticos de una sola región anatómica o de una localización extraganglionar única [Ie] |
| II | Afectación de los ganglios linfáticos de dos o más regiones anatómicas en el mismo lado del diafragma, o de una única localización extraganglionar y uno o más grupos de ganglios linfáticos en e1 mismo lado del diafragma [IIe] |
| III | Afectación de los ganglios linfáticos de regiones anatómicas situadas en ambos lados del diafragma, o de una única localización extraganglionar y de ganglios linfáticos situados al otro lado o de ambos lados del diafragma [IIIe] |
| IV | Afectación difusa o diseminada de una o más localizaciones extraganglionares, con o sin afectación de los ganglios linfáticos |
La presencia de fiebre, sudoración nocturna, o pérdida no justificada por otras causas del 10 % o más del peso corporal en 6 meses, se designa con el sufijo B. La letra A, indica ausencia de estos síntomas. El bazo se considera localización ganglionar a efectos del estadio; la afectación esplénica puede señalarse con el subíndice S, siguiendo al estadio y al estado sintomático
De la modificación de Ann Arbor de la clasificación de Rye.
Los estudios no cruentos que se utilizan para la clasificación clínica por estadios son: Exploración fisica, radiografía de tórax, estudio isotópico de hígado, bazo y hueso, linfangiografía, TAC, ecografía y en algunos pacientes es necesario practicar una biopsia por punción-aspiración medular.
La laparatomía exploradora es muy importante cuando los hallazgos que puedan encontrarse modifiquen de manera significativa la terapéutica y cuando la probabilidad de que esto suceda compense el riesgo y el retraso en el tratamiento que se producen. Los pacientes con una enfermedad de Hodgkin avanzada, incluidos en el estadio clínico IV o en un estadio clínico III poco favorable, deben ser tratados con quimioterapia.
La laparotomía en ellos no está indicada.
En determinados pacientes en el estadio I y II supradiafragmáticos, la probabilidad de encontrar afectación visceral en la laparotomía es lo bastente baja para que sea preferible un tratamiento con radioterapia sin necesidad de laparotomía exploradora.
Los pacientes en un estadio clínico III favorable, o en un estadio I o II no favorable, y con buen estado general, son candidatos a la laparotomía exploradora.
Los estudios que deben practicarse para valorar los estadios de la enfermeded de Hodgkin y en los otros linfomas se resumen en la tabla siguiente.
Pruebas aconsejadas para determinar el estadio clínico y anatomopatológico de la enfermedad
| Enfermedad de Hodgkin | |
| Exploración física/historia | |
| Analítica habitual / hemograma | |
| Radiografía de tórax (PA y lateral) | |
| Linfografía | |
| Estudio isotópico de hígado y bazo | |
| Estudio isotópico óseo | |
| Aspirado y biopsia de médula ósea para los pacientes | |
| – en estadio III | |
| – con síntomas B | |
| – con tipo histológico de celularidad mixta o depleción linfocitaria | |
| – con anomalías hematológicas no justificadas por otra causa | |
| – con fosfatasa alcalina elevada | |
| Estudios opcionales: Gammagrafía con galio radíactivo, tomografía del pulmón, | |
| pielografía IV, ecografía abdominal, TAC de abdomen, biopsia hepática percutánea, | |
| biopsia hepática por laparoscopia. | |
| Laparotomía exploradora en pacientes | |
| – con estadio clínico IIIA, IIB o IB | |
| – con estadio clínico IA o IIA, tipo histológico de celularidad mixta o depleción linfocitaria | |
| – con estadio IA o IIA, con afectación supraclavicular izquierda o del área infradiafragmática | |
| Linfomas no-Hodgkin | |
| Exploración física / historia | |
| Analítica habitual/hemograma | |
| Radiografía de tórax (PA y lateral) | |
| Tomografía computada de abdomen, linfangiografía o ambas | |
| Estudio isotópico de hígado y bazo | |
| Estudio isotópico óseo | |
| Aspiración y biopsia de médula ósea | |
| Estudios opcionales: Gammagrafía con galio radiactivo, tomografía de pulmón, ecografía abdominal, biopsia hepática percutánea, estudio radiológico con contraste de esófago, estómago e intestino delgado, pielografía IV, colangiografía IV | |
La laparotomía exploradora comprende una esplenectomía, biopsia en cuña y dos biopsias profundas con aguja del hígado, así como muestras de los ganglios linfáticos retroperitoneales.
El cirujano debe conocer previamente la linfangiografía y tomar biopsia de todos aquellos ganglios que hayan resultado sospechosos.
Tras la laparotomía se hará una nueva radiografía para comprobar que todos los ganglios sospechosos han sido extirpados.
En otros pacientes puede ser conveniente una biopsia de medula ósea.
TRATAMIENTO
Los estadios I y II de la enfermedad de Hodgkin se tratan de forma efectiva únicamente con radioterapia; el pronóstico es algo peor cuando se acompañan de síntomas B, masas mediastínicas grandes y quizás extensión extraganglionar (IE o IIE]. Los pacientes con un estadio IIIA tienen una probabilidad del 50 al 75 % aproximadamente de sobrevivir libres de enfermedad de 5 a 10 años, cuando se les trata sólo con radioterapia.
En los pacientes en el estadio IIIA2, la terapéutica combinada ha dado buenos resultados. pero también lo ha hecho la quimioterapia aislada.
Los pacientes en los estadios IIIB o IV deben ser tratados con quimioterapia.
Cuando se trata la enfermedad de Hodgkin con intención de curar, la radioterapia debe administrarse en un campo extenso, incluyendo todas las zonas afectadas y los ganglios linfáticos vecinos.
Cuando la enfermedad es supradiafragmática. el campo habitual será el «manto»,
que incluye los ganglios hiliares del pulmón, mediastínicos, infraclaviculares, supraclaviculares, cervicales y axilares.
Se irradia también, en casos apropiados, el anillo de Waldeyer, el parénquima pulmonar vecino a las masas hiliares o mediastínicas y las estructuras infradiafragmáticas.
El campo básico para la enfermedad infradiafragmática, es la «Y invertida», que abarca el pedículo esplénico, los ganglios retroperitoneales, ilíacos e inguinales, y en casos apropiados, el bazo y los ganglios femorales.
Una dosis tipo, podría ser de 4.000 a 4.400 rads, administrados en 4 a 5 semanas, en un campo tipo manto o Y invertida. Se aplican escudos protectores para evitar la radiotoxicidad.
La quimioterapia combinada es muy útil como tratamiento inicial en los estadios IIIB o IV, el protocolo standard es MOPP o COPP (Mostaza nitrogenada, Oncovín, Procarbazina, Prednisona. La mostaza puede ser reemplazada por Ciclofosfamida), basado en ciclos de 28 días, seis ciclos, o un esquema de dos ciclos más, después de obtener una respuesta completa.
Existe otro esquema, ABVD, que se puede alternar: Adriamicina (Daunorrubicina), Bleomicina, Vinblastina y Dacarbacina.
La toxicidad es la propia de cada una de las drogas antineoplásicas, como: náuseas, vómitos, malestar general, alteraciones neurológicas y hematopoyéticas, alopecía, sindrome cushingoide, inmunosupresión, etc.
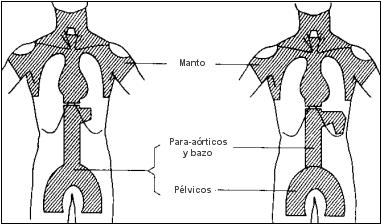
Campos en manto y en Y invertida para la irradiación del sistema linfoide
en la enfermedad de Hodgkin y en los linfomas
LINFOMAS NO-HODGKIN
ETIOLOGIA
Se asocia el linfoma de Burkitt, al virus de Epstein-Barr. Otros linfomas relacionan su aparición a inmunodeficiencias, como las inducidas para el transplante renal, a enfermedades autoinmunes como el síndrome de Sjögren, a la inmunoestimulación crónica, como la que se produce en la enfermedad inflamatoria intestinal y al tratamiento con drogas, como la fenilhidantoina. Se los ha vinculado también a enfermedades no malignas del sistema linfoplasmocitario, como la linfoadenopatía angioinmunoblástica. Por otro lado el Sarcoma de Kaposi conlleva un aumento del riesgo de linfoma.
HISTOLOGIA
El ganglio o la médula ósea infiltrados por un linfoma, muestran la sustitución de sus células y arquitecturas normales, por una población monomorfa y ocasionalmente dimorfa de células; esta infiltración puede ser nodular o difusa.
No se encuentran células de Reed-Sternberg. Algunos linfomas presentan un infiltrado al que se ha llamado histiocítico, debido a la presencia de grandes células
de núcleo vesiculoso y cromatina laxa, pero estos elementos son, por lo general, de estirpe linfoide.
La clasificación de los linfomas está evolucionando en la actualidad. Los estudios inmunológicos de las células linfoides malignas han añadido nuevas perspectivas a la clasificación y al concepto de estos tumores.
La clasificación tradicional en linfosarcoma, reticulosarcoma y linfoma folicular
gigante es vaga y de escaso valor terapéutico o pronóstico, por lo que ya no se utiliza. La clasificación de Rappaport, es sencilla y clínicamente útil. Divide los linfomas de acuerdo con su arquitectura (nodular o difuso) y con su diferenciación citológica.
Clasificación de los linfomas no-Hodgkin según Rappaport
|
Histología |
Pronóstico |
|
| Nodular | ||
| Linfocítico bien diferenciado (NLBD) |
F |
|
| Linfocítico pobremente diferenciado (NLPD) |
F |
|
| Mixto (linfocítico-histiocítico) (NM) |
F |
|
| Histiocítico (NH) |
D |
|
| Difuso | ||
| Linfocítico bien diferenciado (DLBD) |
F |
|
| Linfocítico pobremente diferenciado (DLPD) |
D |
|
| Mixto (linfocítico-histiocítico) (DM) |
D |
|
| Histiocítico (DH) |
D |
|
| Indiferenciado (DI) | ||
|
D |
|
|
D |
|
| Linfoblástico* | ||
| Convoluto |
D |
|
| No convoluto |
D |
|
F. favorable; D. desfavorable
*Primitivemente clasificado como difuso linfocítico pobremente diferenciado
Los estudios inmunológicos hacen pensar en que la mayor parte de los linfomas se originan a partir de células B, que se encuentran normalmente en los folículos de los ganglios linfáticos, el bazo y las amígdalas, así como en los cordones medulares de los ganglios linfáticos, en la médula y en la sangre.
Pocos linfomas, como la micosis fungoide, se originan en los linfocitos T, responsables de la inmunidad celular, que se encuentran en el timo, pulpa blanca esplénica, regiones paracorticales de los ganglios linfáticos, sangre y médula.
Hay una minoría de linfomas que no presentan características de células B ni de células T, a los que se ha denominado linfomas de células nulas.
Clasificación de los linfomas no Hodgkin
|
NCI |
Rappaport o convencional |
| Bajo grado | |
|
DLBD |
|
NLPD |
|
NM |
| Grado intermedio | |
|
NH |
|
DLPD |
|
DM |
|
DH |
| Alto grado | |
|
DH |
|
Linfoblástico |
|
DI incluido el linfoma de Burkitt |
En esta clasificación el pronóstico empeora en sentido descendente (de la A a la J)
Características clínicas
Los linfomas aparecen por igual en ambos sexos.
Aunque muestran predilección por el grupo de edad de 40-60 años, pueden presentarse a cualquier edad.
Al igual que la enfermedad de Hodgkin, suelen debutar con adenopatías periféricas, siendo las más frecuentes las cervicales y supraclaviculares (60 %), inguinales (20 %) y axilares (14%)
También pueden localizarse a nivel central, siendo las masas hiliares y mediastínicas más raras que en la enfermedad de Hodgkin, excepto en el caso de una masa mediastínica anterior en un varón joven, presentación típica del linfoma linfoblástico, la esplenomegalia y las adenopatías abdominales son, por el contrario, formas de presentación comunes.
La afectación predominantemente visceral es más frecuente en los linfomas que en la enfermedad de Hodgkin, encontrándose enfermedad primaria extraganglionar en el 25 % de los pacientes, y enfermedad diseminada visceral en el 50 %.
Las formas extraganglionares más frecuentes son: la del anillo de Waldeyer, con síntomas de masa tumoral o de dolor.
La gastrointestinal, que debuta como hemorragia, dolor u obstrucción.
Otras localizaciones primarias son: la ósea, pulmonar, testicular, tiroidea, cerebral y dérmica.
La diseminación a las visceras suele afectar al hígado, médula ósea y hueso. La primera aparece generalmente como una hepatomegalia.
La infiltración de las meninges no es rara, pero no suele ser una manifestación de comienzo.
Pueden existir síntomas generales, tales como pérdida de peso, fiebre sin causa aparente, y sudoración nocturna (síntomas B). Rara vez una anemia hemolítica auto-inmune o las manifestaciones de la hipercalcemia marcan el inicio de la enfermedad.
Cuando el linfoma no recibe tratamiento, su historia natural varía con el estadio y con el patrón histológico; en las formas de evolución desfavorable se observa caquexia, ascitis y edemas; puede haber insuficiencia renal, en general más por obstrucción ureteral que por infiltración del parénquima.
En algunos pacientes con linfomas de histología favorable, la enfermedad se mantiene indolente durante años, aún sin tratamiento, mientras que en otros avanza progresivamente.
Cuando la histología es desfavorable, el linfoma se disemina rápidamente cuando no se trata y la supervivencia esperada antes de existir métodos terapéuticos eficaces era menor de un año.
Analítica
Las pruebas de laboratorio dan resultados inespecíficos: puede aparecer hiperuricemia cuando la masa tumoral es grande.
Las elevaciones de la fosfatasa alcalina, bilirrubina, SGOT y SGPT pueden indicar afectación de áreas extraganglionares; la anemia hemolítica por anticuerpos o por hiperesplenismo, puede ocurrir.
A veces se ve hipogammaglobulinemia, particularmente en los linfomas tipo B. Cuando se afecta la medula ósea, se encuentran pancitopenias, células linfomatosas circulantes o una fórmula hemática leucoeritroblástica.
Determinación del estadio El estadio suele asignarse en los linfomas del mismo modo que en la enfermedad de Hodgkin.
Los métodos son similares, aunque la tomografía computarizada y la ecografía de abdomen suelen ser más útiles, a causa de la mayor frecuencia de afectación mesentérica u otro tipo de afectación abdominal.
La aspiración y biopsia medulares son también más importantes en estos casos, ya que la médula ósea se ve afectada con frecuencia, y en ese caso el paciente queda inmediatamente clasificado en el estadio IV.
En los linfomas, se puede asignar razonablemente un tratamiento en base al estadio clínico, y en la mayoría de pacientes la laparotomía exploradora no es necesaria.
Los linfomas, al igual que la enfermedad de Hodgkin, tienden a diseminarse a los ganglios por contigüidad, sin embargo, muestran mayor tendencia a respetar grupos ganglionares vecinos y extenderse a otros más distales, creando las llamadas «áreas omitidas».
Los linfomas también se diseminan más a las vísceras, probablemente por via hematógena.
TRATAMIENTO
El tratamiento indicado para los estadios I y II con histología favorable y enfermedad supradiafragmática es la radioterapia curativa.
Las técnicas utilizadas y los campos irradiados son similares a los de la enfermedad de Hodgkin.
El procedimiento en los estadios I y II infradiafragmáticos con masa tumoral pequeña, es similar, aunque puede ser necesario irradiar todo el abdomen para abarcar los ganglios mesentéricos.
Los estadios I y II con histología desfavorable presentan un índice de recaídas muy alto cuando el único tratamiento es la radioterapia.
Existen evidencias de que tanto una modalidad de tratamiento combinada em-
pleando radioterapia y después poliquimioterapia, como la poliquimioterapia aislada pueden obtener índices superiores tanto para la supervivencia global como para el intervalo libre de enfermedad.
Los estadios III y IV con histología favorable pueden tratarse de varias formas: la monoterapia con un agente alquilante, como el clorambucilo o la ciclofosfamida, es bien tolerada y habitualmente eficaz. Algunos pacientes. con enfermedad indolente y masa tumoral pequeña pueden ir muy bien incluso sin tratamiento, aunque deben ser cuidadosamente vigilados, comenzando el tratamiento tan pronto como aparezcan las complicaciones o empiece a acelerarse el ritmo de la enfermedad.
Algunos pacientes con masa tumoral pequeña y bajo grado, son tratados mediante inmunomoduladores tales como Interferón, que en ocasiones se asocia en los esquemas a corticoides y quimioterapia con Leukerán.
Cuando se observa una masa tumoral grande o un tumor de crecimiento rápido o cuando los síntomas son graves, puede aplicarse una terapéutica combinada tipo CVP (ciclofosfamida, vincristina y prednisona), para conseguir la remisión clínica.
Los estadios III y IV con histología desfavorable casi nunca mejoran cuando se tratan sólo con radioterapia o con quimioterapia de agente único, por lo que debe aplicarse una poliquimioterapia apoyada en doxorrubicina y un agente alquilante (por ej., CHOP, Ciclofosfamida, hidroxildaunorrubicina, Oncovin, Prednisona). Aunque existen grupos de mal pronóstico (gran volumen tumoral, afectación medular, afectación meníngea, afectación visceral abdominal), la tasa global de respuestas completas es del 60 al 70%, y la supervivencia libre de síntomas a los 2 y 5 años es significativa (30 a 65%).
Debe agregarse que actualmente se cuenta con una nueva opción terapéutica, especialmente indicada en los estadios III y IV con histología desfavorable, en caso de contar con dadores histocompatibles, esta opción lo constituye el transplante medular, que requiere previamente una esterilización de la médula ósea del receptor, estando este período gravado por una alta incidencia de enfermedades intercurrentes, especialmente infecciosas. Debe atenderse además, una vez establecido el homoinjerto, la posibilidad de un rechazo precoz o tardío, o aún respuesta inmune del material del donante en contra de los tejidos del huesped; no descartándose por otra parte la posibilidad de recidiva de la enfermedad primaria.
OTROS LINFOMAS
El linfoma de Burkitt es poco frecuente y afecta sobre todo a los nìños, pero también puede verse en adultos. Parece encontrarse en relación con el virus de Epstein-Barr, que pudiera ser su causa, y está formado por células B del centro germinal. Suele aparecer en la mandíbula o en el abdomen y se extiende facilmente a la médula ósea, las meninges o las vísceras. Crece muy rápidamente, con un tiempo de duplicación de 72 horas, y es muy sensible a la quimioterapia. Cuando se administra ciclofosfamida (con o sin vincristina, prednisona y methotrexate), se produce una gran necrosis tumoral, que puede acompañarse de complicaciones metabólicas graves y potencialmente letales si la masa tumoral es grande. Su pronóstico depende del estadio y las supervivencias a largo plazo sin sintomatología varían entre el 20 y el 70 %.
Cuando aparece afectación meníngea, medular o abdominal inoperable, el pronóstico es muy malo. La media de curaciones es aproximadamente del 55 %. Cuando aparece una masa abdominal importante, la resección quirúrgica mejora el pronóstico.
La micosis fungoide es un linfoma cutáneo de células T.
Los linfomas linfoblásticos suelen ser neoplasias de células T que debutan como masa mediastínica anterior, y de medula ósea. Suelen encontrarse en varones adolescentes, pero pueden aparecer también en mujeres. La afectación meningea puede aparecer tardiamente. El pronóstico es malo El tratamiento con la poliquimioterapia compleja utilizada en las leucemias agudas linfoblásticas es preferible al tratamíento habitual de los linfomas. Debe hacerse también profilaxis meníngea.
El linfoma de Lennert se caracteriza por la gran cantidad de histiocitos epitelioides que presenta; suelen ser linfomas de células T de histología desfavorable.
En el tratamiento de estos casos se utiliza el protocolo CHOP.(ciclofosfamida, hidroxildaunorrubicina, Oncovin, Prednisona)
El linfoma mediterraneo es un linfoma multifocal del intestino delgado. Se encuentra sobre todo en los paises de Oriente Medio y en las costas mediterráneas. Suele haber un antecedente de gammapatía monoclonal, la enfermedad de las cadenas alfa, que se caracteriza por diarrea, malabsorción, infiltración plasmocitaria del intestino delgado, acropaquias y aparición en suero de una proteína monoclonal.
El pronóstico es muy malo y no se ha encontrado hasta la fecha la terapéutica adecuada.
La linfadenopatía angioinmunoblástica con disproteinemia, caracterizado por adenopatías generalizadas que habitualmente se acompaña de hepatosplenomegalia. Se observa anemia hemolítica con prueba de Coombs positiva, fiebre, erupción cutánea, y una gammapatía monoclonal.
Los ganglios linfáticos muestran: proliferación vascular, infiltrado polimorfo de linfocitos, células plasmáticas, inmunoblastos y eosinófilos; en el intersticio se deposita un material eosinófilo.
Se piensa que representa una proliferación de células B, con una regulación T anómala. Suele evolucionar hacia un linfoma (sarcoma inmunoblástico).